|
|
GRAM-NEGATIVE OUTER MEMBRANE MODELER
Automated building of Lipopolysaccharide-rich Bacterial Outer Membranes
|
Force Fields
GNOMM currently supports the following force fields: | |
CHARMM (Chemistry at Harvard Macromolecular Mechanics) is a widely used set of all-atom force fields for Molecular Dynamics simulations. The CHARMM Development Project involves a worldwide network of developers working with Martin Karplus and his group at Harvard to develop and maintain the CHARMM program. The current implementation of the force field (CHARMM36) includes parameters for proteins, nucleic acids, carbohydrates, lipids and various types of solvent. At the same time, the CHARMM General Force Field (CGenFF) offers an extensive selection of chemical groups present in biomolecules and drug-like molecules, enabling the generation of parameters for a wide range of organic ligands and compounds. |
GROMOS (GROningen MOlecular Simulation) is a family of
united-atom force fields, i.e. without explicit aliphatic (non-polar) hydrogens. GROMOS has been developed since 1978 for the dynamic modelling of (bio)molecules by the research group of Wilfred
van Gunsteren at the University of Groningen, The Netherlands until 1990, and since then at the ETH, the Swiss Federal Institute of Technology, in Zürich, Switzerland. In GROMOS, polar hydrogens are
modeled explicitly, while aliphatic and aromatic hydrogen atoms are included implicitly by representing the carbon atom and attached hydrogen atoms as one group centered on the carbon atom. The force
field has been optimized with respect to the condensed phase properties of alkanes and includes parameters for proteins, lipids and many organic compounds. |
MARTINI is a coarse-grain (CG) force field suited for molecular dynamics simulations of biomolecular systems. Developed by Marrink and coworkers at the University of Groningen, Martini was initially designed for simulations of lipids and was later extended to various other molecules, including proteins, carbohydrates, inorganic polymers and nucleic acids. The force field has been parametrized in a systematic way, combining top-down and bottum-up strategies: Non-bonded interactions are based on the reproduction of experimental partitioning free energies between polar and apolar phases of a large number of chemical compounds, whereas bonded interactions are derived from reference all-atom simulations. The model uses a four-to-one mapping, i.e. on average four heavy atoms and associated hydrogens are represented by a single interaction center. In order to keep the model simple, only four main types of interaction sites are defined: polar, non-polar, apolar, and charged. Each particle type has a number of subtypes, which allow for an accurate representation of the chemical nature of the underlying atomistic structure. |
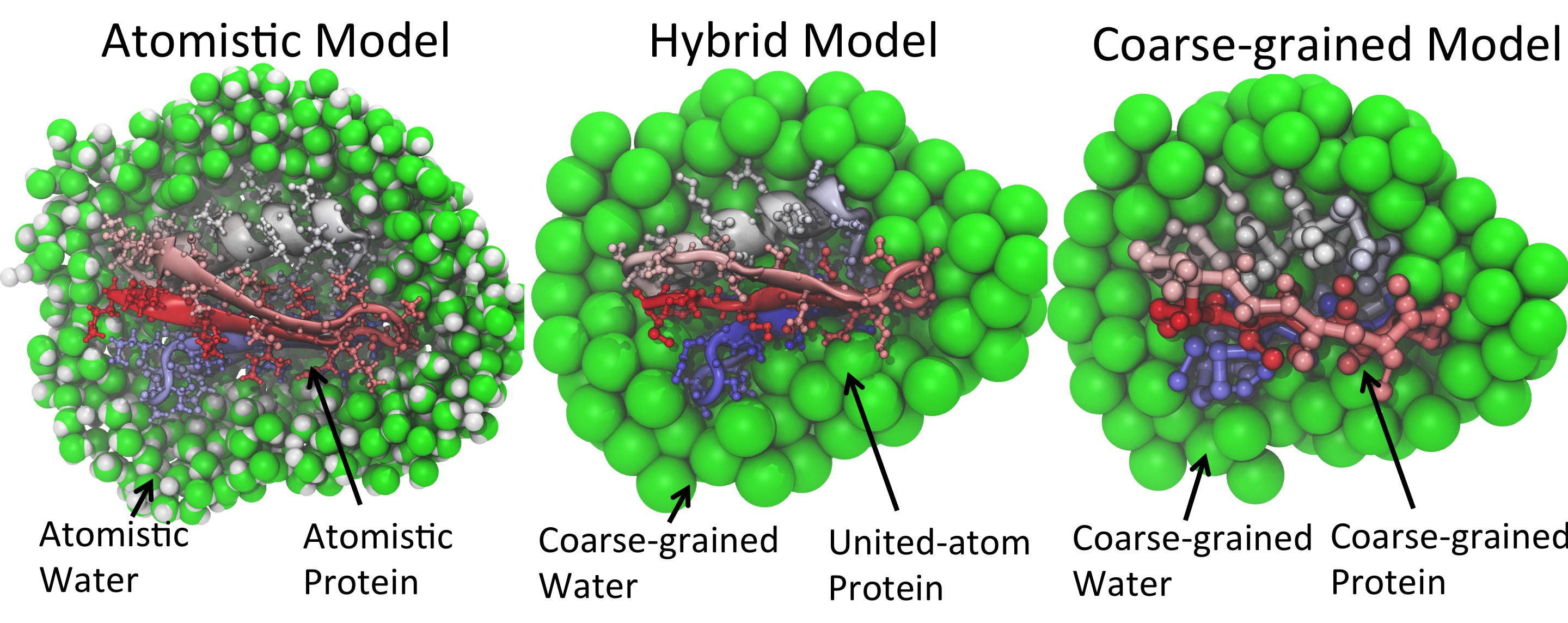 | |
PACE is a novel, hybrid approach on force field development for biomolecular simulations, utilizing a combination of different resolutions for modeling different parts of the system. Proteins are modeled using a United-Atom (UA) representation, while the lipids and solvent use the MARTINI coarse-grain (CG) force field. Initially developed by Han and coworkers and later extended by the Klaus Schulten group at the Theoretical and Computational Biophysics Group at the University of Illinois at Urbana-Champaign, PACE has been successfully used in protein folding simulations, the study of amyloid fibrils and the simulation of transmembrane proteins, including both transmembrane alpha-helices and beta-barrels. The model uses a GROMOS-like potential energy function to describe the proteins, complemented by additional energy terms to correctly reproduce the directionality of hydrogen bonds in alpha-helices and beta strands, the standard MARTINI energy function to model the CG environment, and additional, tabulated non-bonded terms for the cross-interactions of UA and CG particles. The latter have been parametrized by comparing partition-free energy values to experimental evidence on small organic molecules. |
Fotis A. Baltoumas, Stavros J. Hamodrakas & Vassiliki A. Iconomidou (2019)
The Gram-Negative Outer Membrane Modeler: automated building of lipopolysaccharide-rich bacterial outer membranes in four force fields
J. Comput. Chem.
Jul 5; 40(18): 1727-1734, DOI: 10.1002/jcc.25823
Make sure JavaScript is enabled in your browser to achieve full functionality.