|
|
GRAM-NEGATIVE OUTER MEMBRANE MODELER
Automated building of Lipopolysaccharide-rich Bacterial Outer Membranes
|
Manual
What is GNOMM?
The Outer Membranes of Gram-negative bacteria are asymmetric lipid bilayers, composed by standard lipids in the inner leaflet and mostly lipopolysaccharides (LPSs) in their outer leaflet. Outer membranes and their components are of critical importance for bacterial cell structure and function, as outer membrane proteins can regulate a diverse range of functions, while the organization and dynamics of LPSs can influence bacterial resistance against antimicrobial agents and cause toxic reactions in human hosts, leading to a number of diseases.
Despite their significance, the experimental study of outer membranes is challenging. Molecular Dynamics (MD) simulations can be a useful tool in modeling the structure and dynamics of membranes and membrane proteins. However, the preparation and simulation of outer membrane proteins in their native LPS-containing outer membrane environment is not straightforward.
The Gram-Negative Outer Membrane Modeler (GNOMM) is an automated workflow for the construction of LPS-rich outer membrane systems in MD simulations. GNOMM currently supports four widely used force fields, namely, the CHARMM36 all-atom, GROMOS 54A7 united-atom, MARTINI coarse-grained and PACE hybrid resolution models, and enables the building of membrane and protein-membrane systems with complex lipid bilayers containing LPSs. The generated output configurations can be subsequently used to perform Molecular Dynamics simulations with the freely available, high performance GROMACS simulation engine.
Availability
GNOMM is publicly available through http://bioinformatics.biol.uoa.gr/GNOMM.
Usage
Users can start using GNOMM through the Submit page. The submission form is presented in Fig. 1:
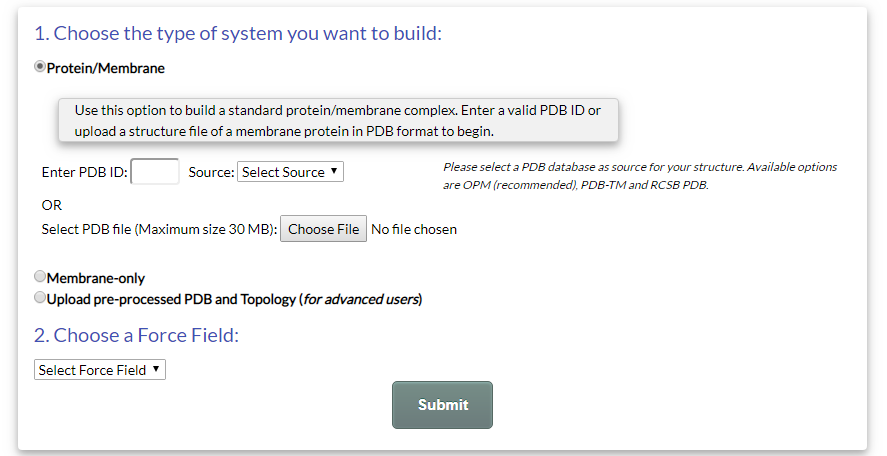
Figure 1.The Submission Form of GNOMM
GNOMM currently supports the creation of three different types of simulation systems:
- Protein-Membrane System: Users can submit the PDB file of a protein structure and build a protein/membrane system. This is the default option of GNOMM.
- Membrane-only System: Users can simply build a lipid bilayer.
- Pre-processed PDB and Topology System: Users can submit a pre-processed structure (in PDB format) and topology (in GROMACS top/itp format) and build a membrane system around it. This option is suitable for cases where the simulation system requires the presence of "non-standard" elements, i.e. molecules or particles that are not part of the default force field topology. For example, these can include:
- Protein modifications (e.g. phosphorylation, cross-linking modifications) or non-standard aminoacids (e.g. selenocysteine) that are not, by default, part of a force field.
- Non-protein coordinates, e.g. a ligand (such as an antibiotic) that is bound to a protein. In this case, the user must derive the force field topology of the molecule and provide it alongside the protein.
- Inorganic polymers, such as fullerenes or carbon nanotubes. Such structures are not part of traditional biomolecular force fields and their topology must be built by the user.
The following manual sections will present the modeling procedure of GNOMM in a step-by-step basis.
1. Building a Protein/Membrane system
To build a "Protein/Membrane" system, select the option "Protein/Membrane" in the Submit page (Fig. 2).
To proceed with the submission, you can either enter a valid PDB ID or upload a structure file in PDB format (note that, for efficiency, the maximum size of PDB uploads is currently 30 MBs). Note that the structure should be pre-oriented with respect to the membrane plane. This means that the PDB's Z- principal axis needs to be aligned to the membrane plane's normal. Such files can be retrieved from the Orientations of Proteins in the Membrane (OPM) database or the Protein Data Bank of Transmembrane Proteins (PDB-TM), or can be generated through the PPM server. Using RCSB PDB as the source of input, although available, is generally NOT recommended since the structures are not likely to have the proper orientation.
- If you choose to enter a valid (i.e. four characters) PDB ID, you should also choose the database from which it can be retrieved. Three databases are currently supported:
- OPM: The Orientations of Proteins in Membranes (OPM) database contains pre-oriented coordinate files for transmembrane, peripheral and lipid-anchored proteins.
- PDB-TM: The PDB-TM database contains pre-oriented coordinate files for transmembrane proteins.
- RCSB: The RCSB PDB is the default PDB archive and contains the original structures as deposited by their respective authors. Note that these structures may not have the proper orientation with respect to the membrane plane. Therefore, the use of RCSB PDB, although available, is not recommended.
- If you choose to upload your own PDB file, note that it must have the proper orientation, as described above. Such files can be generated using the PPM Server available in OPM.
An example of a submission is shown in Fig. 2.
In this example, the structure of OmpA (PDB:1BXW) is chosen, with OPM as the source database. The force field is set to "CHARMM36".
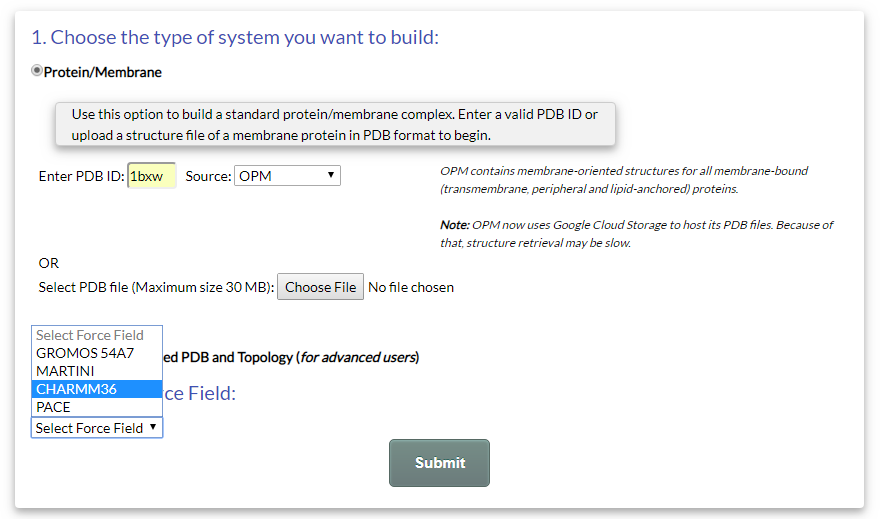
Figure 2.Example submission of a "Protein/Membrane" system for OmpA (PDB: 1BXW), retrieved from OPM.
If no errors occur, you will be redirected to the system setup page. Otherwise, an error message will appear and you will be prompted to go back and change your submission.
The system setup page is divided into three sub-section, corresponding to the three main steps followed by the GNOMM modeling process:
- Protein Preparation: Definition of protein topology features.
- Membrane Definition: Definition of membrane size and composition
- Solvation and Ionization: Definition of the solvent properties
Step 1. Protein Preparation
The first section (Fig. 3) includes the following fields:- Job ID: Upon submission, each job receives a unique numerical ID. You can use this ID to track your job throughout the building process and retrieve your results.
- Structure manipulation: The structure manipulation interface provides two features:
- A structure viewer (LiteMol), which you can use to inspect your uploaded structure.
- A number of force field related options. These are dependent on the force field you have chosen to use. By default, you are asked to define if any modifications should be made regarding the charge state of the protein's N- and C-termini. Depending on the force field, some other options may also be available. Specifically, each force field has the following options:
- CHARMM36: Charge state of termini.
- GROMOS 54A7: Charge state of termini.
- MARTINI: Choice of force field version (either standard MARTINI or MARTINI-ELNEDYN) and charge state of termini.
- PACE: Choice of force field version (1.3-1.5) and charge state of termini.
- PDB structures may have a number of errors, including but not limited to: missing or incomplete coordinates, non-standard atoms, erroneous atom or residue names. These errors may cause your job to fail during generation of the protein topology. GNOMM can fix these errors using MODELLER.
Note that by default, GNOMM can only fix simple mistakes such as those referenced above. If your structure has larger parts of the protein missing (e.g. loops or entire domains), you should consider constructing a model through homology modeling prior to using GNOMM.
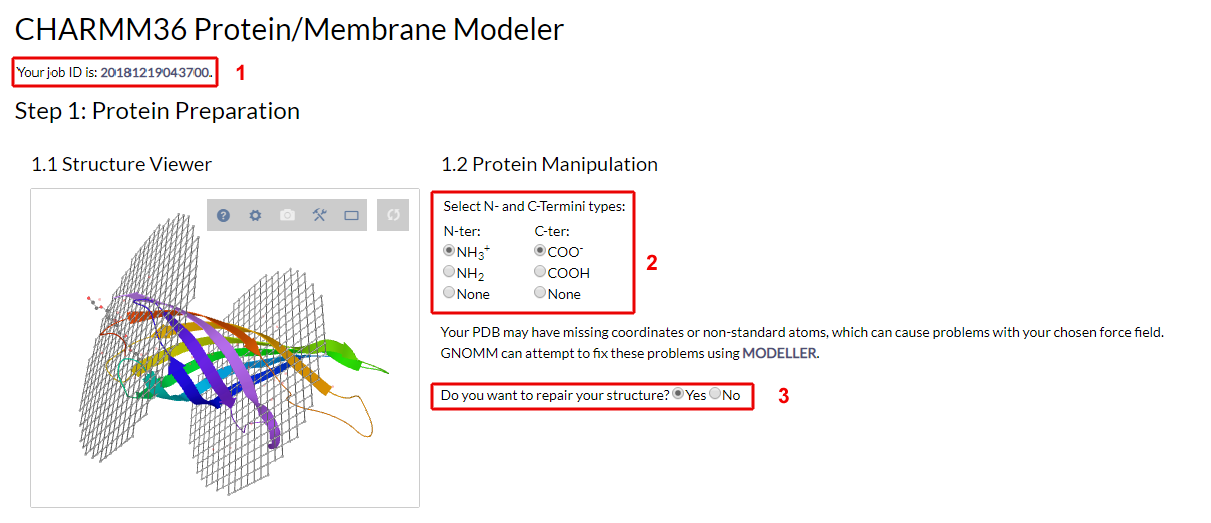
Figure 3. The protein preparation form
Step 2. Membrane Definition
After defining the protein's features, you move on to defining your membrane's size and properties. The first step is defining the X and Y dimensions of the lipid bilayer (Fig. 4):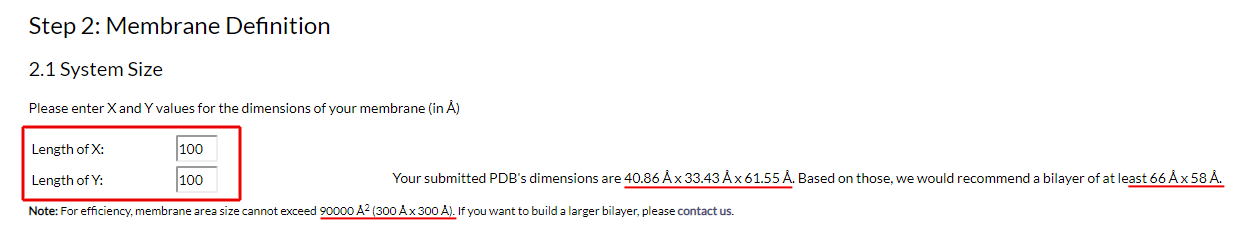
Figure 4. Definition of membrane dimensions
Use the form, as shown in Fig. 4, to enter the X and Y values of the bilayer's dimensions. Make sure that these are large enough to include your protein and leave ample space for the packing of lipids, and to properly respect periodic boundary conditions. To aid you, GNOMM calculates the unit cell dimensions of your submitted protein and, based on those, provides the minimum suggested values for you to enter.
Note that X and Y values may not necessarily be the same. You can enter different values and produce non-square membrane patches if you need.
Also note that for computational efficiency, the membrane surface area must not exceed 90000 Å2.
After you have defined the membrane's dimensions, you need to define its composition, by filling out the table shown in Fig. 5:
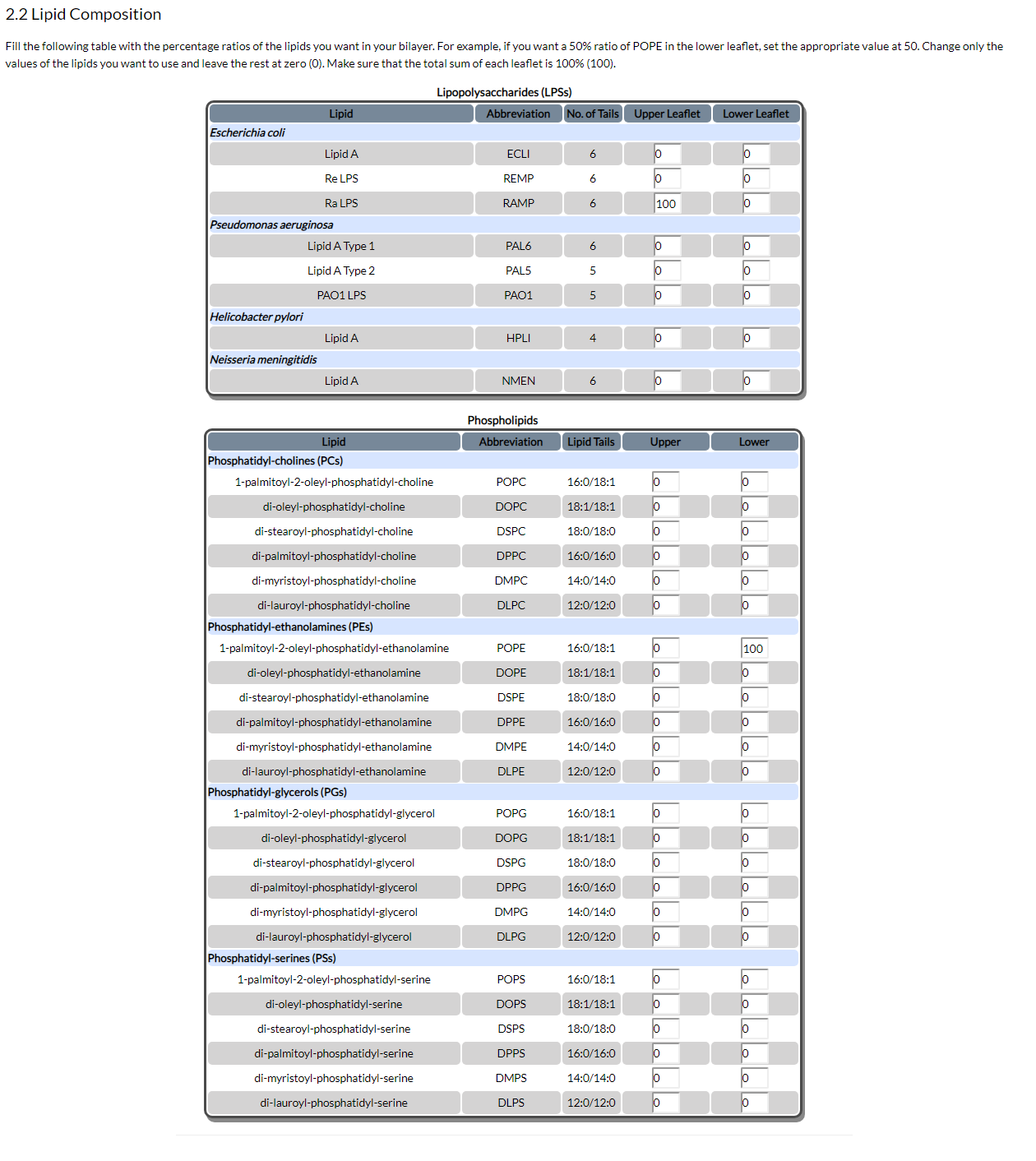
Figure 5. Definition of membrane composition
Fill the table with the percentage ratios you want in your table. Change only the values of the lipids you actually want to use, leave the rest at zero (0). Make sure that the total sum of each leaflet is 100% (100).
In the figure's example we want a bilayer with a uniform Ra LPS (RAMP) upper leaflet and a uniform POPE lower leaflet. Therefore, we enter 100 for RAMP in the upper leaflet and 100 for POPE in the lower leaflet.
Step 3. Solvation and Ionization
In step 3 (Fig. 6) you choose whether you want to add solvent layers on each side of your system and whether you want to add ions:
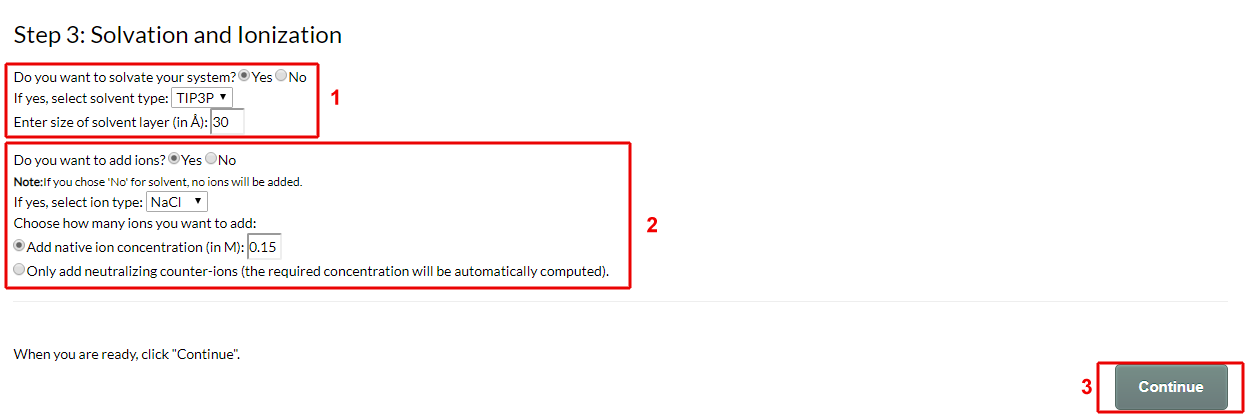
Figure 6. Solvation and ionization
- Choose if you want to solvate your system. If yes, choose the water model you want to use and define the size (i.e. height) of the solvent layer on each size of the membrane.
The available solvent models for each force field are TIP3P or TIP4P for CHARMM, SPC or SPC/E for GROMOS, Standard CG or Polarizable CG for MARTINI, and Standard CG for PACE. - Next, choose if you want to add ions. If yes, select the ion type (NaCl, KCl or CaCl2) and define its concentration. Two options are available:
- Add custom concentration: add a native ion concentration of your choice. Default value is 0.15 M.
- Only add neutralizing counter-ions: Add as many ions as needed to neutralize your system's charge. In this option the concentration of ions will be automatically determined by GNOMM.
- When you are ready, click the "Continue" button to begin building your system.
Job Progression and Results
After your job is submitted, you will be redirected to the following page (Fig. 7):
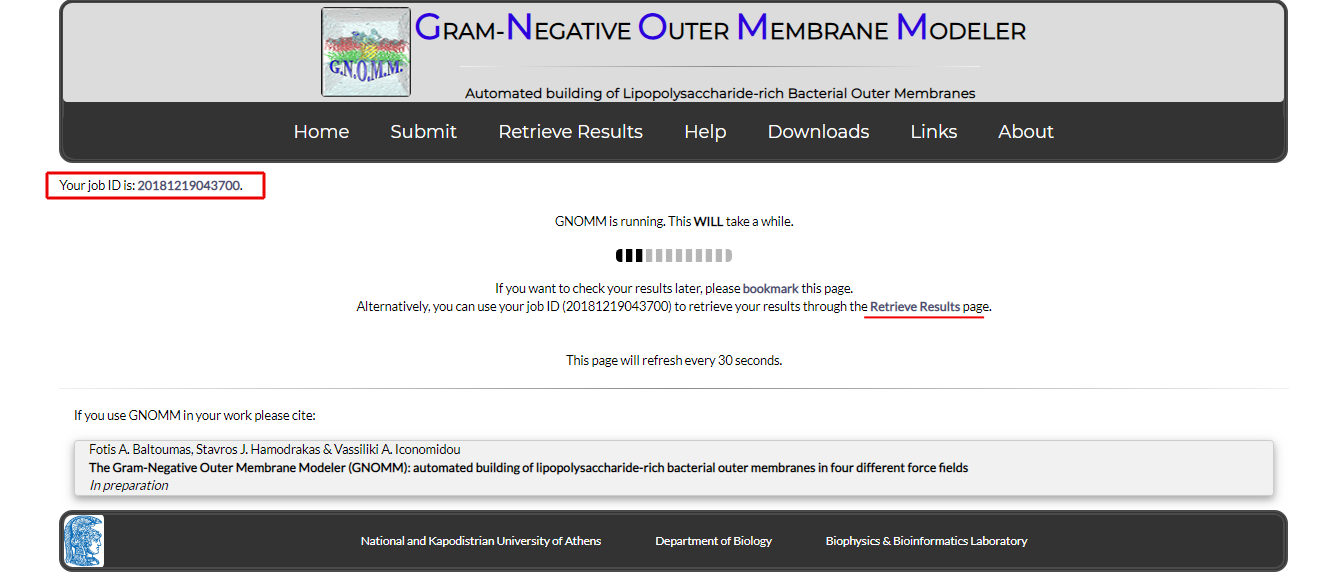
Figure 7. The job process page
Since building may take a while, you are strongly advised to bookmark this page and return later. Alternatively, you can use your job ID to check your status through the "Retrieve Results" form (see the relevant section below).
When your job is complete, you will see the following page (Fig. 8):
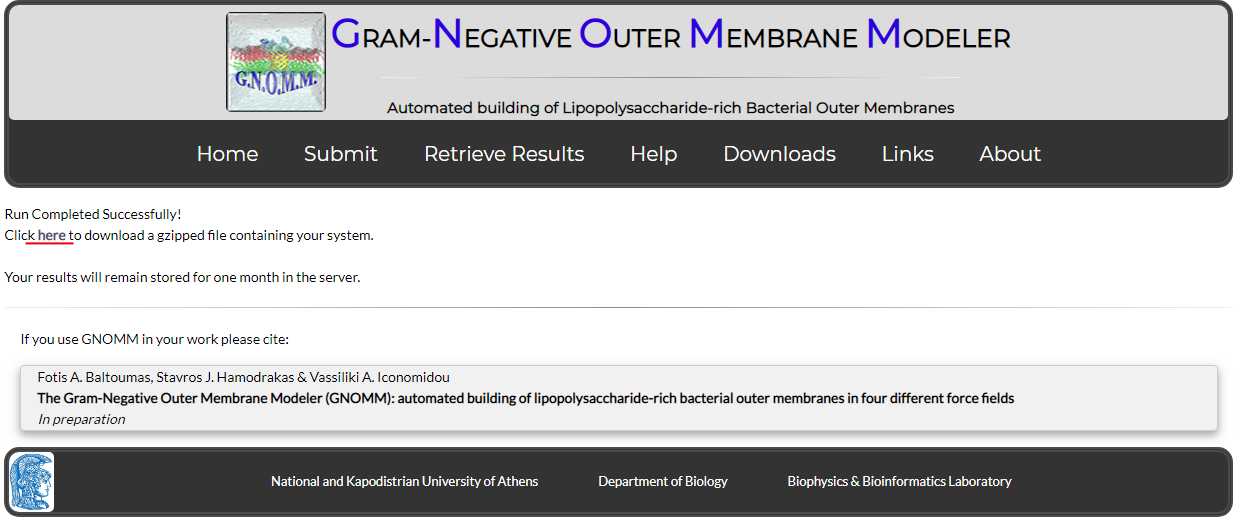
Figure 8. The results page
Click the provided link to download a compressed file with all your results (Fig. 9). These will include the following:
- The structure of your complete system in PDB format ("final.pdb");
- The topology file ("topol.top") and, depending on your system, a number of individual component topologies (itp files) describing your system.
- A directory containing all required force field files (e.g. "charmm.ff") to prepare a simulation.
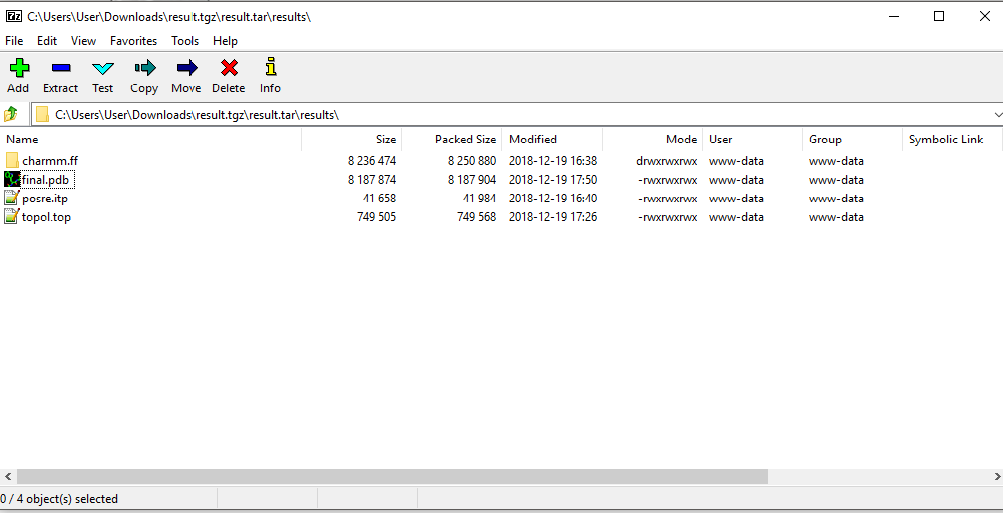
Figure 9. Contents of the results
Note that your results will remain stored in the GNOMM server for 30 days.
Job Retrieval
You can retrieve the results of your previous jobs or check the progress of a currently running job by using the form in the "Retrieve Results" page. In the form, just input the ID corresponding to the job you submitted.
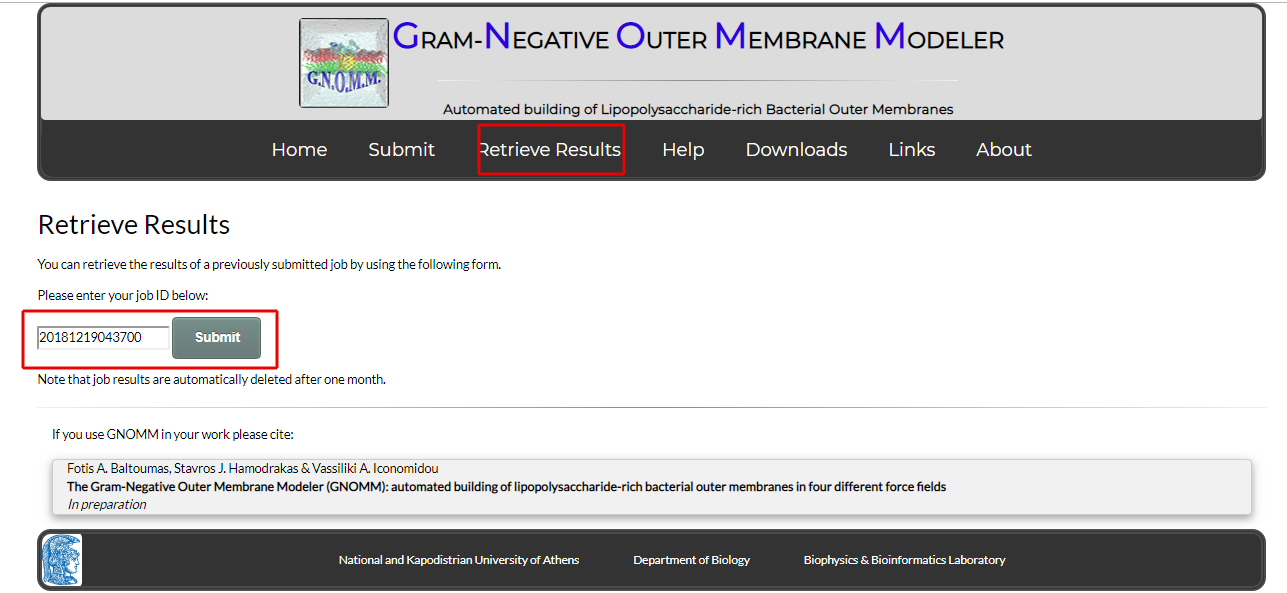
Figure 10. The Retrieve Results page
2. Building a Membrane-only system
To build a "Membrane-only" system, simply select the option "Membrane-only" in the Submit page, choose a force field and click the "Submit" button (Fig. 11):
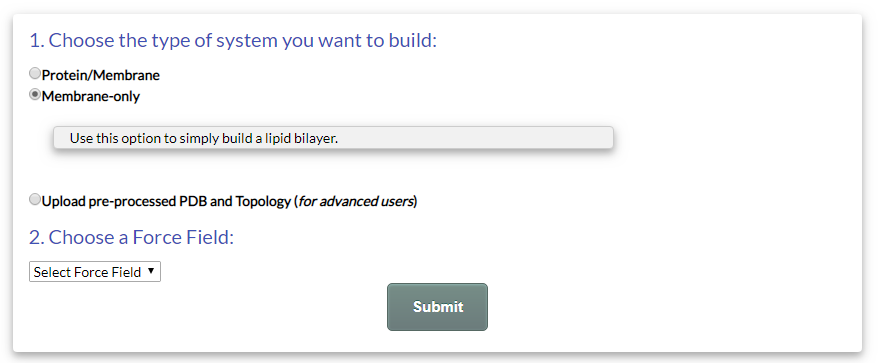
Figure 11. The "Membrane-only" option in the Submit page.
Since you are building a Membrane-only system, the first part of the GNOMM modeling process (protein manipulation) is skipped. The rest of the process (membrane building, solvation and ionization) is *exactly* the same as in the Protein-Membrane builder interface.
3. Building a system with a pre-processed PDB and topology
To build a system using a pre-processed PDB structure and its acompanying topology, select the "Upload pre-processed PDB and topology (for advanced users)" option in the Submit page (Fig. 12):
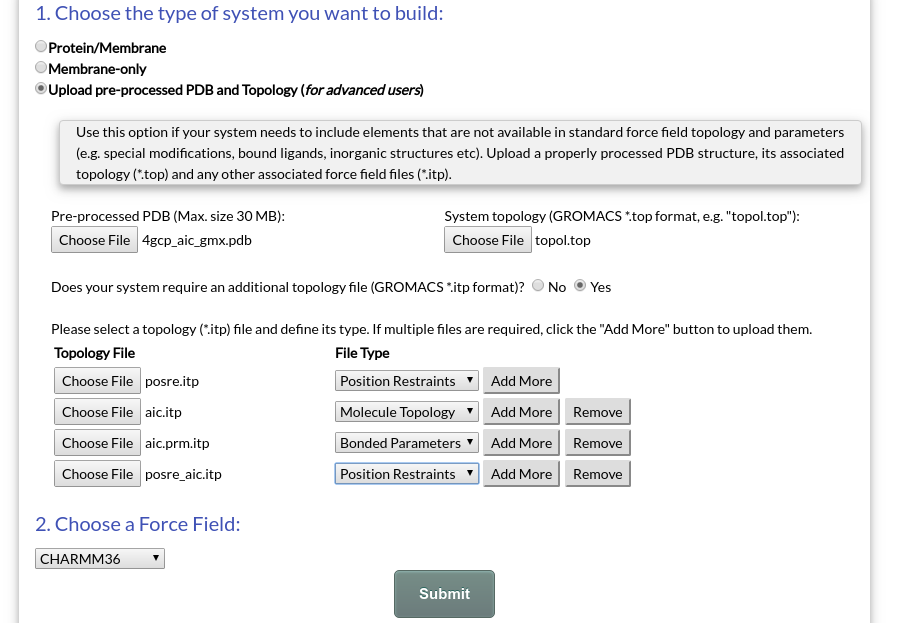
Figure 12. Example submission of a system with a pre-processed PDB structure and topology.
In the example of Fig. 12, we prepare a simulation for OmpF in complex with ampicillin (AIC), an antibiotic (PDB: 4GCP). The protein has been pre-processed for use with GROMACS using pdb2gmx and the CHARMM36 force field. For AIC, force field parameters have been generated using CGenFF.
Note that the process of building a protein-ligand system for simulations requires an intermediate (at least) knowledge of how GROMACS works. There is an excellent tutorial in Prof. Justin Lemkul's GROMACS Tutorial site (www.mdtutorials.com/gmx/) for the construction of protein-ligand systems (see here). You are advised to consult this tutorial and any other relevant material to familiarize yourself with the subject.
Fotis A. Baltoumas, Stavros J. Hamodrakas & Vassiliki A. Iconomidou (2019)
The Gram-Negative Outer Membrane Modeler: automated building of lipopolysaccharide-rich bacterial outer membranes in four force fields
J. Comput. Chem.
Jul 5; 40(18): 1727-1734, DOI: 10.1002/jcc.25823
Make sure JavaScript is enabled in your browser to achieve full functionality.